Transcription
The Martin Lab has a long history of studying fundamental mechanisms in transcription. Using powerful tools of biophysical chemistry and enzymology, we have focused on the (relatively) simple, single subunit RNA polymerase from bacteriophage T7. Our work is all with purified enzyme and largely with synthetic DNA templates, affording us exquisite control over the system. The story below represents insights from our lab, but of course, also from a wide variety of excellent labs, in both the T7 and multi-subunit RNA polymerase families [citations not yet added ... soon?].
While this is written from the perspective of the T7 family of single subunit enzymes, most of it also holds true for the multi-subunit RNA polymerases. While the multi-subunit bacterial and eukaryotic RNA polymerases clearly have a common evolutionary ancestor, the single subunit polymerases have almost certainly evolved independently. That many features of transcription are the same or similar across these families recognizes the common requirements that the process has imposed on evolutionary selection.
T7 RNA polymerase is an ideal model system for studying fundamental mechanisms in transcription. There are two broad classes of DNA-dependent RNA polymerases: 1) the multi-subunit polymerases, of which the bacterial RNA polymerase and eukaryotic Pol II are best known and 2) the single subunit RNA polymerases, of which T7 RNA polymerase and the mitochondrial (and chloroplast) RNA polymerases are best known. The single subunit enzymes are related to the large Pol I family of DNA and RNA polymerases. Although these two broad classes are evolutionarily distinct, the big picture mechanisms are a case of convergent evolution – similarities reflect the constraints of the complex challenge in evolving an enzyme that is initially very sequence-specific, but transitions to an enzyme that can transcribe any sequence.
T7 RNA polymerase is widely used to synthesize RNA in vitro, from a DNA template. The single subunit RNA polymerase from bacteriophage T7 was initially characterized in 1973, but with its cloning and over expression in 1984, it became a work horse for the in vitro synthesis of RNA of any length. The Uhlenbeck lab pioneered this application (and early on, described some limitations!). With the success of the SARS-CoV-2 vaccines, this use has been widely recognized and the Nobel Prize in Physiology or Medicine was awarded in 2023 to researchers who pioneered its use with modified bases. Coming from a bacteriophage with a 40 kb dsDNA genome, the enzyme has been used in vitro to generate RNA as long as 27 kb!
Some general words about transcription
Unlike a textbook enzyme where substrate binds, catalysis occurs, and product is released, a polymerase must move along the DNA, with repeated cycles of catalysis/movement (the primary job of a DNA polymerase). An RNA polymerase maintains a short (about 8 base pair) RNA-DNA hybrid, within a melted bubble in the DNA, but then releases RNA beyond (upstream, away from the active site) that point into solution. The DNA to which the RNA, and the substrate NTPs, bind is called the template strand - it provides the encoding information. The DNA displaced in the bubble is the nontemplate strand and is actually not needed in vitro (except at the promoter).
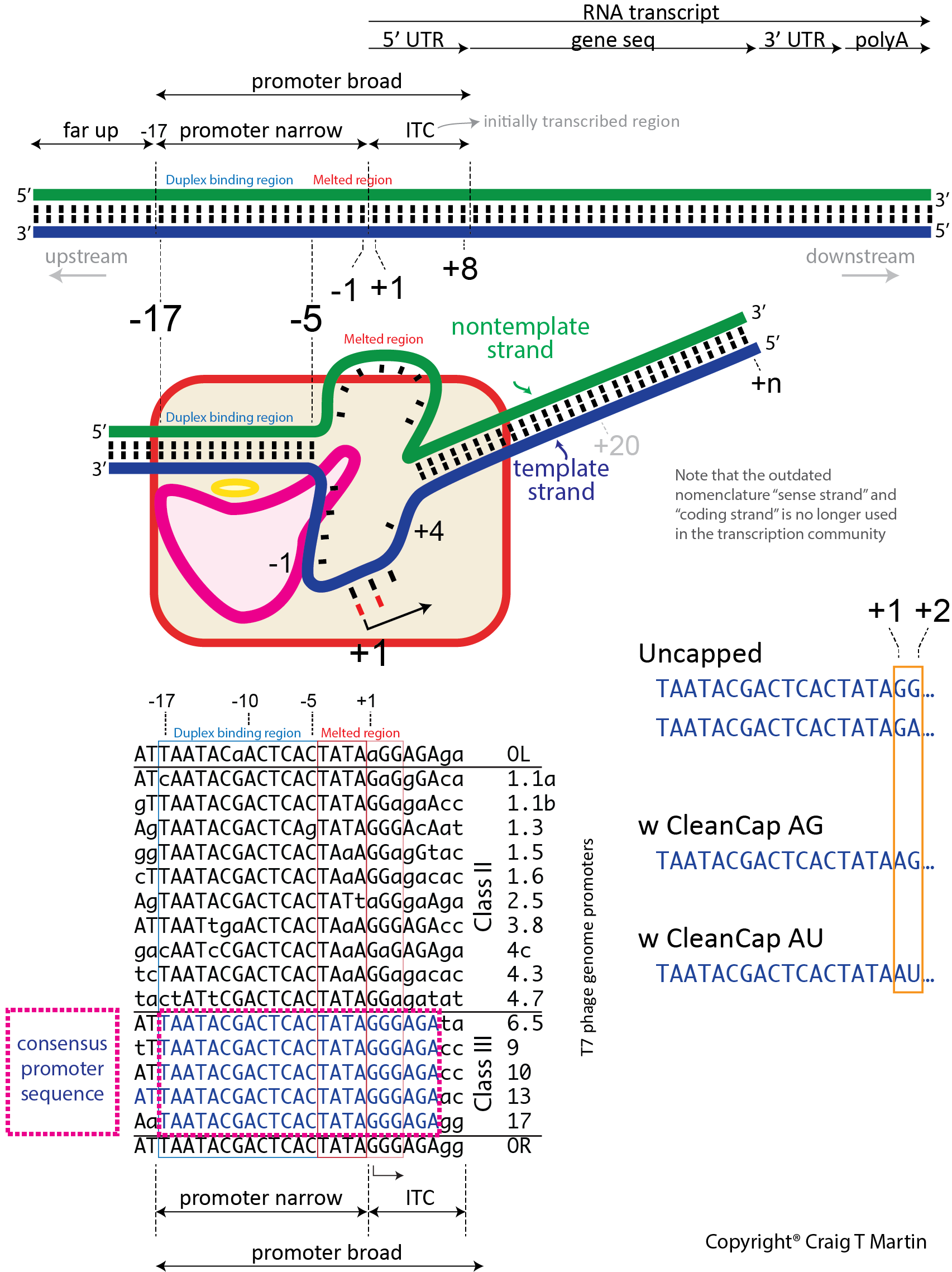
de novo initiation. Unlike a DNA polymerase, an RNA polymerase must initiate transcription de novo - that is, without a pre-formed primer. And it must do so at specific sequences (promoters) in the DNA. Thus, T7 RNA polymerase binds a promoter sequence that extends upstream about 17 bases from the transcription start site. It recognizes features in the duplex region from positions -17 to -5, and melts a "bubble" extending from position -4 to about +3 (transcription starts at position +1, and there is no position 0!). Some of the binding energy from the duplex DNA contacts is used to melt open and maintain this initial bubble. The primary initiation event involves the first two substrate NTPs, sitting down at positions +1 and +2, followed by a phosphoryl transfer reaction (see below) to create the first phosphodiester bond, releasing pyrophosphate from the +2 NTP. The triphosphate from the +1 NTP is retained passively - we call this the 5' end of the transcript. At this point, the polymerase must move forward (translocate) along the DNA (or the DNA move backward within the polymerase), to allow positioning of the +3 NTP in the active site, encoded by the +3 base in the template strand. Another phosphoryl transfer reaction occurs to generate a 3 base transcript (a 3mer RNA).
Dinucleotide initiators, such as Clean CAP skip this first step. In principle, they should initiate with higher affinity than the two separate NTPs above. To further enable this distinction, current dinucleotide initiators are ApG, rather than GpG, since the polymerase initiates less efficiently with ATP.
Initial transcription (leading to abortive RNAs). To translocate again, on duplex DNA, the downstream end of the bubble must melt to expose the base at +4 in the template DNA. Since RNA polymerase retains its duplex promoter interactions, the upstream end of the DNA bubble remains fixed and so the bubble is now expanding (forward). Repeated rounds of translocation/phosphoryl transfer extends the RNA and expands the bubble (if this region artificially contains no nontemplate strand, everything still occurs normally, in the absence of a bubble). Interestingly, another thing is happening as the RNA grows during initial transcription: the RNA-DNA hybrid duplex grows from 2, to 3, to 4, etc base pairs. In T7 RNA polymerase, the growth of that hybrid, as the hybrid translocates backwards within the polymerase active site, very quickly causes RNA-DNA hybrid to "bump up" against a domain in the polymerase, pushing on it and causing it to move (translocate) and rotate. Addition of RNA bases at the active site lengthens the hybrid, which serves as a growing piston to drive this motion of the N-terminal domain. This is critical, as energy from phosphoryl transfer is being converted into mechanical "strain" in the form of this piston motion.
Transition to elongation. Why did nature evolve this piston/strain process? To consider this, we need to back up briefly. During the process just described, we have a 2, 3, 4, ... base RNA-DNA duplexes (as the hybrid is growing). Without an enzyme present, the bubble wouldn't form in the first place, but even if it did, it would immediately collapse back down, displacing the newly synthesize 2, 3, 4 base RNA. Remember that the energy of promoter binding was used to melt the bubble and continues to be used to maintain the upstream edge of the bubble. Therefore, during initial transcription, when the RNA-DNA hybrid is short, nature must maintain those strong promoter contacts, to maintain the bubble open. Eventually the hybrid becomes long enough (see below) to resist collapse of the bubble, and at this point, the promoter contacts can be lost. Indeed, at this point, it is important to release the duplex promoter contacts, so that the enzyme can move 10, 100, 1000 bases downstream. The promoter contacts are strong - we need an input of energy to drive release of those contacts, but the only favorable energy input in the system derives from the phosphoryl transfer reaction. The growing RNA-DNA hybrid piston is the mechanical/energetic coupling nature needs. Its growth drives a structural change that ultimately leads to weakening promoter contacts - and promoter release. The use of the growing hybrid as a piston has independently evolved (at least) twice in evolution. Although the structural details are very different, the multi-subunit RNA polymerase in bacteria and eukaryotes also uses the hybrid as a mechanistic piston to drive promoter release.
Elongation. In both T7 RNA polymerase and the multi-subunit RNA polymerases, the enzyme maintains an ≈8 base pair RNA-DNA hybrid. Why 8 base pairs? It has been proposed that 8 base pairs is sufficient for the RNA (red) to be topologically locked (helically) around the DNA template (blue) strand. It is that topological threading that confers the extreme stability of an elongating complex. If the RNA were to transiently un-pair, it is still locked in place, topologically. Note that in the absence of the polymerase active site, a generic 8 base pair RNA-DNA hybrid is not stable in solution.
Note that forward translocation requires 1) melting of one base pair downstream in the DNA, 2) re-annealing of a base pair at the upstream end of the DNA, and 3) melting of one base from the RNA-DNA hybrid. (1) and (3) are expected to be unfavorable, while (2) should be favorable. The net balance is expected to be sequence dependent, such that elongation can be faster or slower than the reported average of 250 cycles per second.
The complete elongation cycle here has been proposed to proceed via a "Brownian ratchet" mechanism. The movement and NTP binding steps are expected to be reversible and so the system is present in a distribution of states reflected by the 2 forward and the 2 reverse rate constants. The bond formation step (phosphoryl transfer) should be irreversible. Thus forward translocation could proceed back and forth, as thermal motion, but when the correct NTP binds, bond formation can occur, locking the complex one step forward. This process repeats throughout RNA synthesis, at an average rate of 250 cycles per second for T7 RNA polymerase.
See Elongation Elaborated below for more on elongation complex stability!
Fidelity. RNA polymerase fidelity can be understood in the context of the dynamic distribution discussed above. While mismatched bases (NTPs) can bind, they presumably populate the bound state for shorter times than correct bases. Therefore, they add at (much) lower rates. It is well-known in the polymerase community that replacing Mg(II) in reaction by Mn(II) increases polymerase error rates. The thought is that since Mn(II) coordination is stronger, it drives binding of both correct and mismatched bases. Since the former already bind well, the effect is to differentially favor the latter. The mismatched base has a longer residence time, which allows for more (mis)incorporation. Might high concentrations of NTPs and Mg(II) have a similar effect?
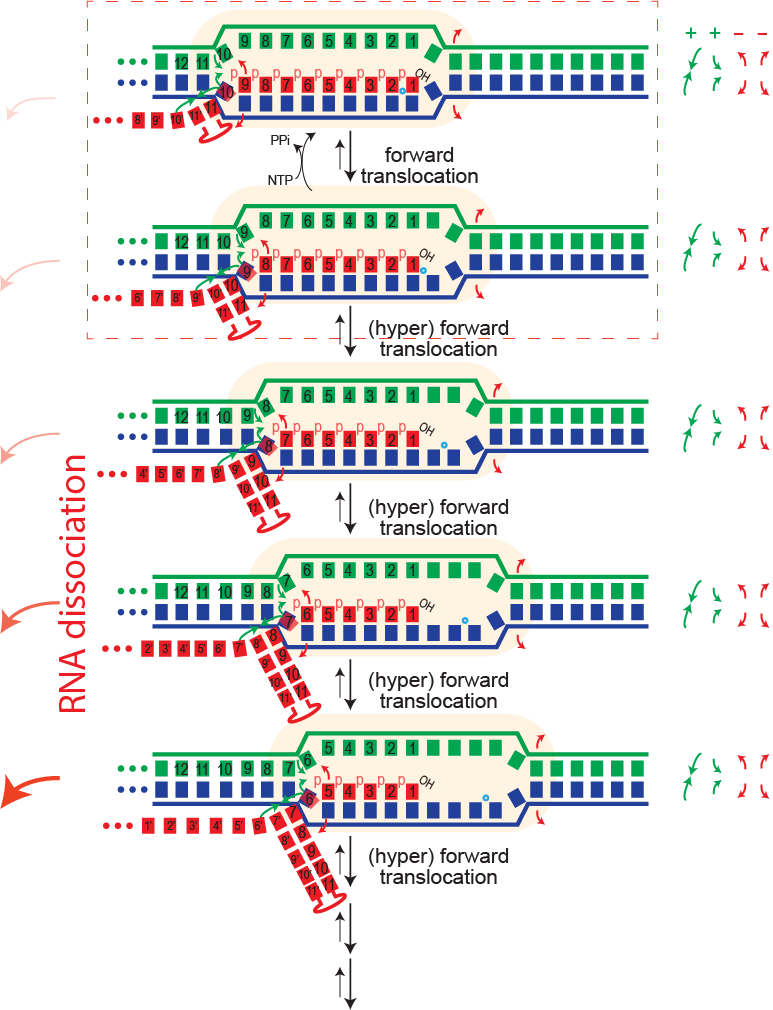
Dissociation/termination. If the topological lock is a major stabilizing factor in elongation, then one must "un-thread" that lock to achieve dissociation of the complex. It is proposed that this happens in hairpin-dependent terminators and in Rho-dependent termination in the bacterial RNA polymerase. In the former case, formation of a hairpin in the exiting RNA "pushes" the polymerase forward (hyper-forward translocation), unwinding the topological lock. In the latter case, an ATP-driven motor protein achieves the same end. In runoff transcription (perhaps rare in biology, but very common in laboratory applications), when the polymerase gets to the end of the DNA, there no downstream DNA duplex and therefore that barrier to hyper-forward translocation is gone. The polymerase slides forward and unthreads the lock, to achieve dissociation.
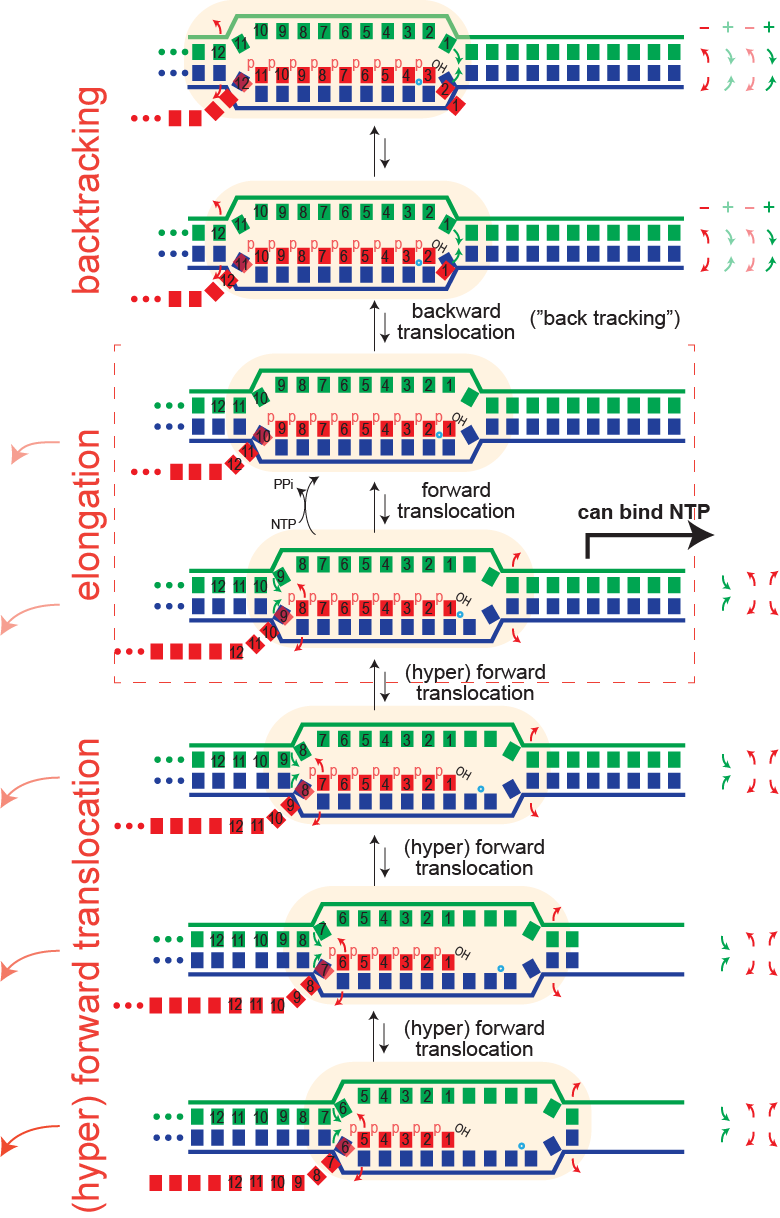
Pausing. Hyper forward translocation removes the 3' hydroxyl of the RNA from the active site. Elongation cannot proceed from hyper forward translocated states, but the complex can transition back to the on pathway stay. So in principle, at least, "shallow" excursions in the forward direction could lead to pausing.
Just as an elongation complex can hyper-forward translocate, it can also reverse translocate (known as "backtracking"). As with hyper forward translocation, the RNA 3' hydroxyl is moved out of the active site and elongation necessarily halts, and as before, transcription is halted until the complex returns to the productive cycle position, with the 3' hydroxyl at the active site. Backtracking has been often observed in multi-subunit RNA polymerase (but has not yet been directly demonstrated in the single subunit (T7) RNA polymerases).
Note that backtracked complexes are expected to be very stable, as they have a full topological lock (one might argue they have an even more complete topological lock than complexes in the normal elongation cycle).
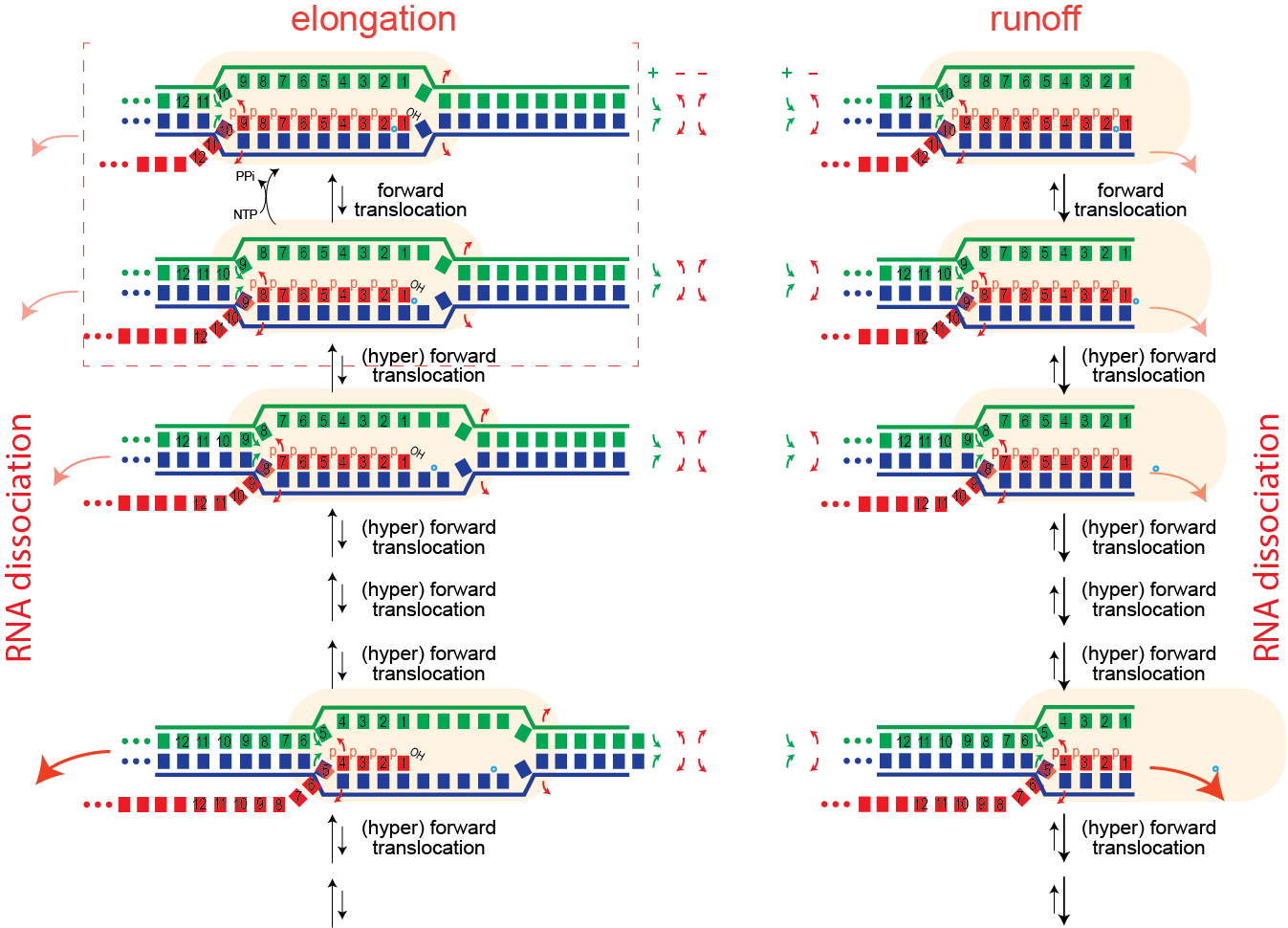
Runoff transcription. If elongation complexes are so stable, how do we achieve high turnover in RNA manufacturing/synthesis in vivo? The above provides an answer. In the normal elongation cycle, hyper forward translocation requires 1) melting of one base pair downstream in the DNA, 2) re-annealing of a base pair at the upstream end of the DNA, and 3) melting of one base from the RNA-DNA hybrid. At runoff, the the first barrier is missing, shifting the energetics more favorably towards the hyper translocated states, and therefore to dissociation. Dissociation at runoff is much faster than internal dissociation, allowing for more cycles of synthesis. This instability as the complex approaches the end of DNA likely leads to n-1 and n-2 products often observed.
Phosphoryl transfer - the key to almost all nucleic acid metabolism. A word about phosphoryl transfer: it's fundamental sophomore organic chemistry and lies at the heart of all polymerases, nucleases, ligases, etc. Polymerases follow a "two metal" mechanism. One stabilizes the pentacoordinate phosphorous intermediate in the central transition state above. The other comes in with the nucleoside triphosphate and then leaves with pyrophosphate. Since Mg(II) coordinates the triphosphate of each NTP in solution, polymerase reactions need more than one Mg(II) per NTP. Note that the free concentration of Mg(II) can be much lower than the total concentration in solution. However, including pyrophosphatase in a reaction, which cleaves pyrophosphate, frees Mg(II), such that the free concentration of Mg(II) increases during the course of a reaction.
Note that during de novo initiation (see above), "Base n" (position +1) above is a single nucleotide triphosphate and so is bound very weakly in the active site. For this reason, Km for the binding of the +1 NTP is weaker than for all of the other NTPs (which all bind at the n+1 site and so have strong binding through their triphosphate groups).
Elongation elaborated - stability. RNA polymerases are motor proteins. As noted above, they "ratchet" along the template strand with some level of forward force. RNA polymerase can be stopped mid-DNA via two general mechanisms (though there are more!): 1) artificially, one can halt an RNA polymerase by starving it of substrate. So for example, if the first encoded C is at position +50, in the presence of ATP, GTP, and UTP (specifically, omitting CTP), RNA polymerase will transcribe to position +49 and wait for the CTP that isn't coming. Alternatively (2), one can create a road block downstream. The most effective roadblock is a covalent crosslink between the nontemplate and template strands, preventing the downstream melting described above. Both of these approaches will halt T7 RNA polymerase. One can also create a road block by binding a protein very tightly to a particular sequence in the RNA. This works well for the multi-subunit polymerases, but T7 RNA polymerase has been shown to displace these binders. It appears to be a better motor. In fact, if a (leading) T7 RNA polymerase is stalled or slowed, a trailing T7 RNA polymerase can displace the leading RNA polymerase, bumping it off the DNA and releasing the truncated RNA.