T7 RNA Polymerase Mechanism and Structure
Jump to sub-topic
- Promoter recognition
- de novo initiation
- Promoter clearance - abortive cycling - transition to elongation
- Elongation complex stability
- Fidelity of transcription
- Bumping
- Termination
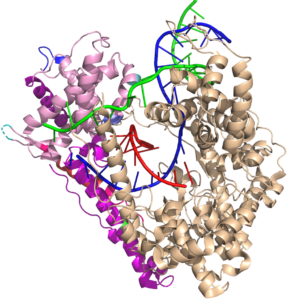
A model system for the central dogma. T7 RNA polymerase is the most well-studied of the single subunit DNA-dependent RNA polymerases. Although it appears to be evolutionarily unrelated to the cellular multi-subunit RNA polymerases, it share many common mechanistic features, suggesting that convergent evolution has solved challenges similarly, at least twice, driven by the demands of transcription
A powerful platform for RNA therapeutics and technologies. T7 RNA polymerase is widely used in vitro (and in vivo) to express RNAs efficiently. Its relative lack of regulation and its relatively simple form (simple for a complex RNA polymerase) makes it ideal for efficient production of mRNA, lncRNA, or just about any RNA. It does have some limitations, which we are working to overcome. More on that elsewhere. Here we offer an overview of a complex molecular machine.
The narrative below describes transcription in T7 RNA polymerase from a Martin Lab success perspective. It is a work in progress - stay tuned for a more complete summary of this important enzyme/process.
The basics
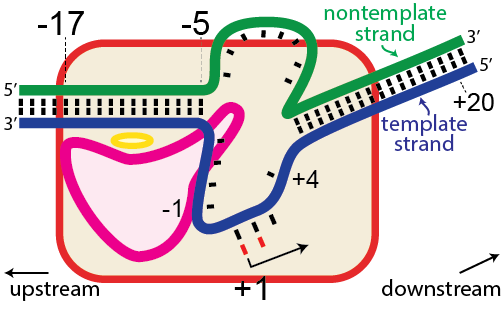
Before proceeding to structure and mechanism, a bit of an introduction to nomenclature.
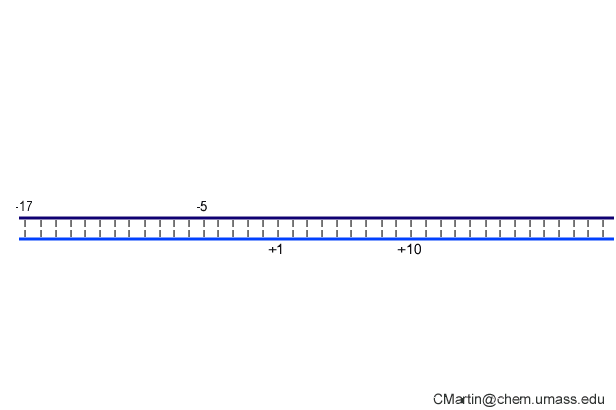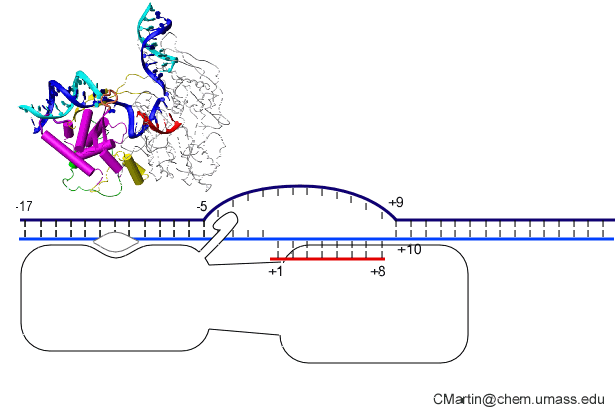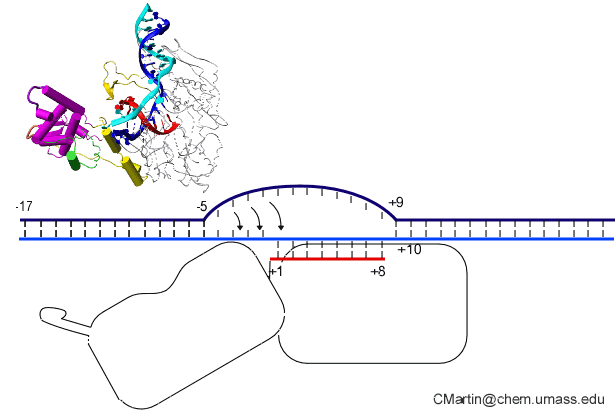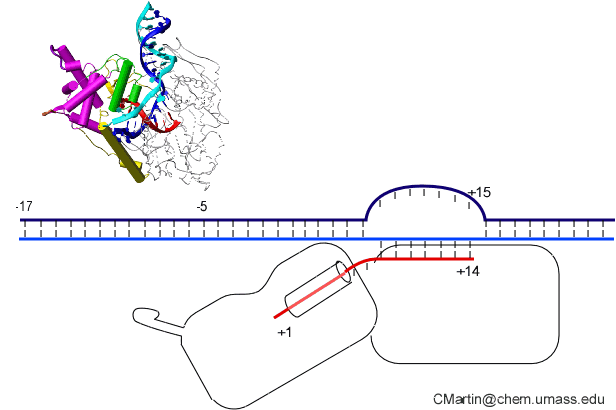
In the numbering of an RNA polymerase promoter, the first base in the DNA to encode nascent RNA is denoted as +1. The direction that the polymerase moves (to the right in the figure here) is called "downstream." The opposite direction (including bases not encoding RNA) is called "upstream" and the numbering goes into negative numbers.
In this numbering system, there is no base "0" - the base at -1 lies immediately upstream of the base at +1.
The DNA strand that serves as a template for RNA is called the template strand. The DNA strand that is not used in templating is called the nontemplate strand. Ignore older nomenclature, which was then and remains now confusing.
The consensus T7 RNA polymerase promoter, as defined by the 17 promoters in the T7 phage genome, extends from position -17 to about position +3, but see below. The cartoon above is shown in parallel with the initiation complex crystal structure below.

Promoter recognition
The consensus promoter sequence is derived from the 17 known promoters in the bacteriophage T7 genome. Class III promoters are generally considered to be the strongest promoters and are the most highly conserved. Thus, the consensus promoter sequence is generally considered to be
TAATACGACTCACTATAGGG
and extends from position -17 to position +3. Different regions of this sequence play different roles in initiating transcription.
As elaborate further below, the bulk of the binding energetics derives from contacts with the duplex region from position -17 to position -5. Of those contacts, the most important region is the major groove of duplex DNA from position -5 to about about -11, with additional interaction with the minor groove from about position -13 to -17.
The first 3 initially encoding bases are generally included in the definition of a promoter. For the consensus sequence, the first three bases are GGG. however, promoters with an initially encoded sequence of GGA also function well.
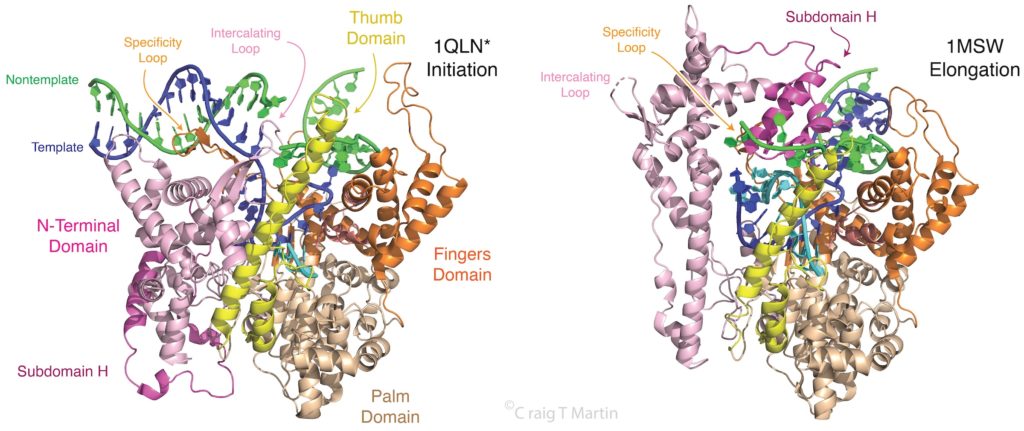
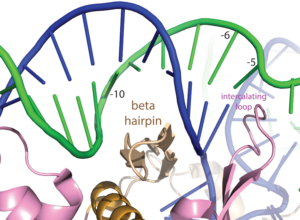
Note that a protein beta hairpin from the C-terminal domain rests on the N-terminal platform and makes contacts with the central major groove from about -7 to -11. Small differences in residues in this hairpin distinguish the T7 RNA polymerase / promoter interaction from its close relatives. The fact that this recognition element comes from a different part of the protein will become important as we discuss the transition to elongation below.
Promoter opening. Some of the favorable energetics of these upstream contacts is used to drive melting of the DNA bases from positions -4 to about +3. In order to maximize the upstream duplex contacts, a beta hairpin in the N-terminal domain is forced (intercalates) into the DNA duplex. Interactions with hydrophobic residues in this intercalating loop stabilize the exposed hydrophobic face of the remaining duplex. As long as upstream promoter contacts are maintained, the intercalating loop is positioned to keep the DNA duplex from collapsing.
de novo Initiation
RNA polymerases, unlike DNA polymerases, can initiate de novo (from the beginning) RNA polymer synthesis from just the first two building blocks (bases). Promoter binding and DNA melting set the stage, but how do the first two bases form a dimer, bound to the template strand? In particular, the enzyme needs to maintain the DNA open until the nascent RNA-DNA hybrid is sufficiently stable to resist the DNA simply collapsing and ejecting the nascent RNA. Maintenance of the intercalating loop, driven by promoter binding, keeps the bubble open.
Promoter Clearance is accompanied by a large change in the protein
Note that in the above structure, there is only room for a 3 bp RNA-DNA hybrid. Yet we know that the elongation complex accommodates an 8 bp hybrid. This means that the protein must adjust its shape (by domain movement) to accommodate a growing hybrid. Put another way, the growing hybrid "pushes" on the N-terminal domain to induce a rotation/translation, all the while maintaining promoter contacts (which, as noted above, keeps the DNA bubble from collapsing and displacing the initial short RNA DNA hybrid).

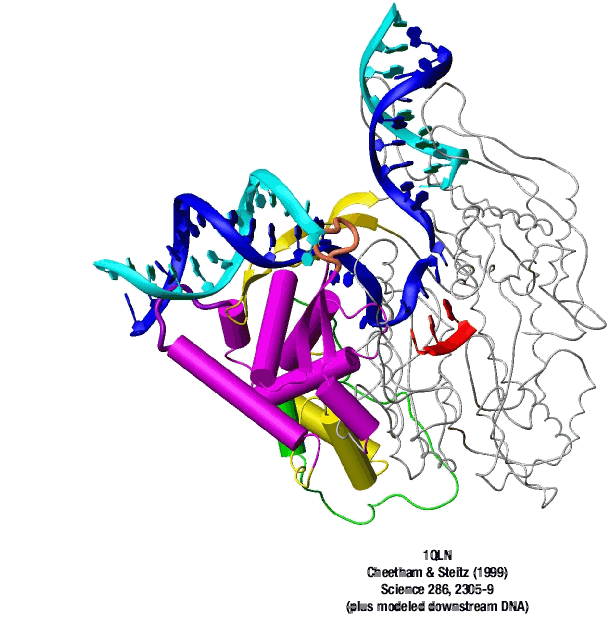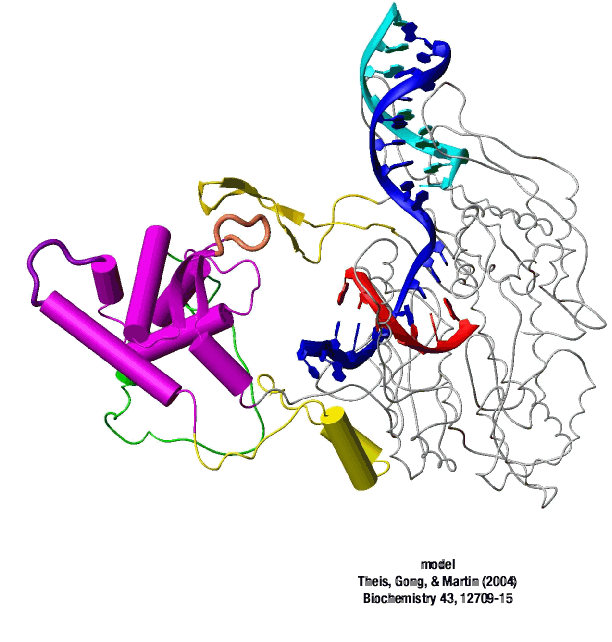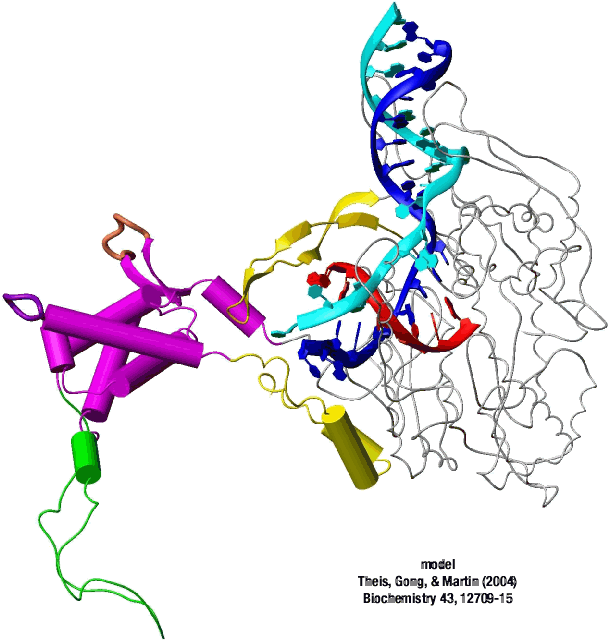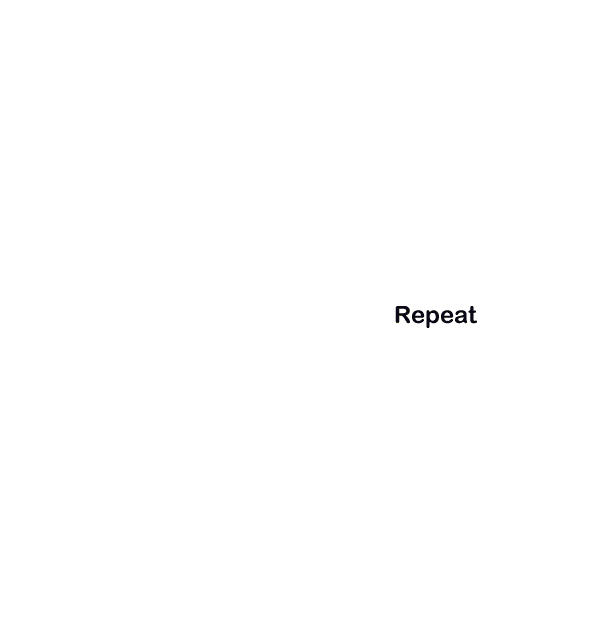
This is seen in the structure of an intermediate (+8) with an 8 bp RNA-DNA hybrid above. This is thought to be at or near the tipping point at which a larger transformation occurs. In what we can define as the transition to elongation, the upstream promoter DNA is released from its contacts, the intercalating loop releases from its interactions with open bubble, and the N-terminal domain then rotates 220° in the direction opposite to the above rotation.
The end result is an N-terminal domain lacking a specificity loop -- the elongation complex can no longer recognize promoter DNA and is now sequence non-specific.
Note in the above that in the elongation conformation (1MSW), the N-terminal platform on which the specificity loop formerly sat is now in the top left corner of the image, illustrating the large rotation. More importantly, a region known as subdomain H (darker violet within the pink N-terminal domain), which is in the lower left corner of the initiation and initially transcribing structures has moved up and to the right, refolded, and now forms a part of the RNA exit channel.
Promoter clearance includes abortive cycling
In initial transcription, the hybrid is very short and so the complex is unstable. Retention of promoter contacts insures some stability. The transition to elongation, however, requires the release of stable promoter contacts. The above provides a nice way to drive the timed release of promoter contacts. In particular, using the RNA-DNA hybrid as a growing piston couples the excess energetics of nucleotide addition to a progressive weakening of promoter contacts, driving promoter release. Disruption of those contacts is maximal at a hybrid length of about 9 base pairs, providing a (positional) timing of release. At 8 base pairs, the complex has established a topological lock (see below).
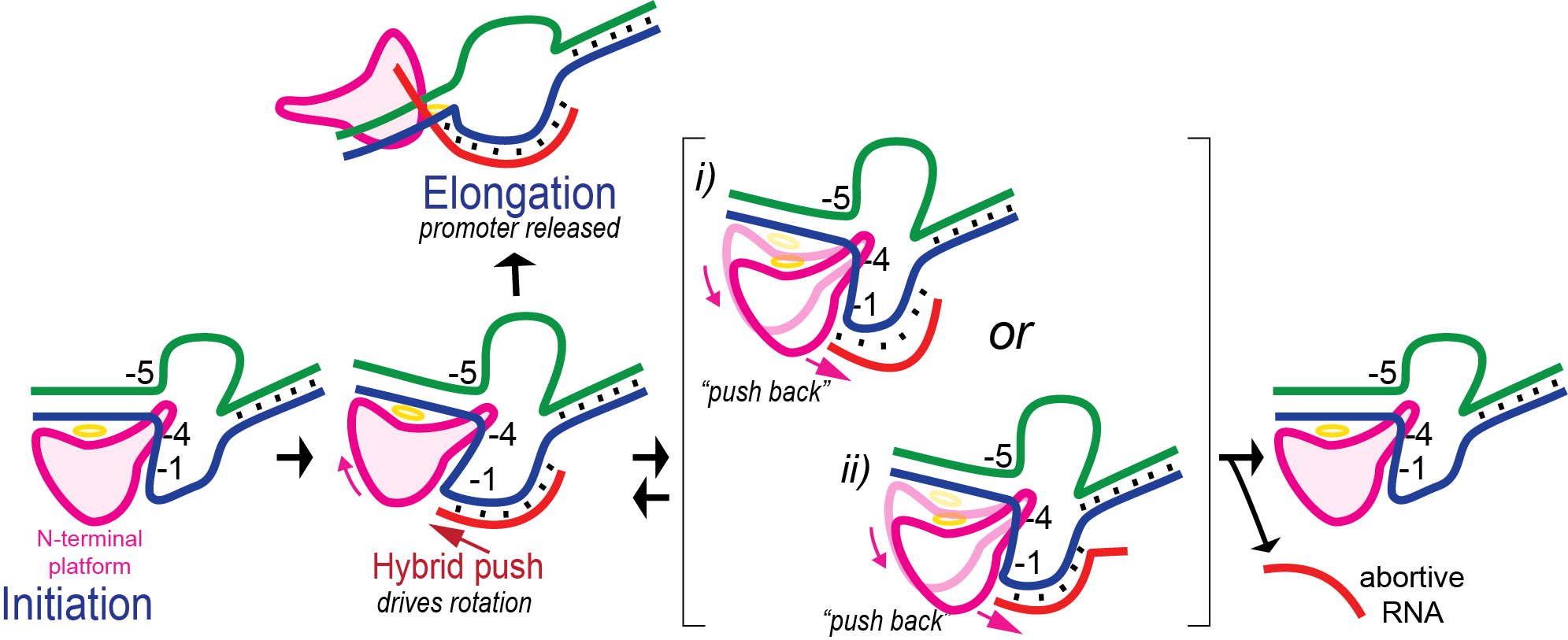
In the above, growth of the hybrid "pushes" against the rotation N-terminal domain. By microscopic reversibility, the N-terminal domain must also "push back" on the hybrid. It is thought that the resulting instability of the hybrid leads some complexes to release a short RNA and return to the initial position - this process is called abortive cycling. Note that promoter contacts are maintained during this cycling (though cycling includes a return to the initially bound complex, which can dissociate).
Various known mutations, reduce the "push back" by the enzyme and lead to significantly fewer abortive products. Those same mutations, however, also lead to some failure to release at the correct translational position, and result in dead end complexes that release RNA of length 11-13 base pairs.
Elongation complex stability
All RNA polymerases have about an 8 base pair RNA DNA hybrid in their elongation complexes; as a base is added at the 3′ end, an upstream base dissociates (resolves) from the hybrid and enters an RNA exit channel, on its way to exiting the protein.
Why 8 base pairs? RNA polymerases, from the small single subunit enzyme here, to much larger and unrelated RNA polymerases from bacteria and humans, all maintain an about 8 base pair RNA-DNA hybrid during elongation. Indeed, initial transcription is designed to stabilize the system until this length is achieved. Why 8 base pairs? It has been hypothesized that 8 base pairs is the length required to establish a topological locking of the RNA around the template strand DNA.
Note in the structure that the red RNA strand, with its 3' end in the active site (rightmost base) and forms an 8 bp RNA-DNA hybrid. The remainder of the 5' end of the RNA exits the polymerase towards the "back" of this structure as shown. As such, the green nontemplate strand of the DNA and blue template strand cannot come together because their reannealing is blocked by the RNA. The transcription bubble is locked open. This is independent (and in addition to) any thermodynamic stability of the RNA-DNA hybrid (which is expected to be only somewhat more stable than the reannealed DNA-DNA duplex).
For this reason, elongation complexes that are stopped in the middle of a DNA stretch are stable for more than an hour. That is unless....
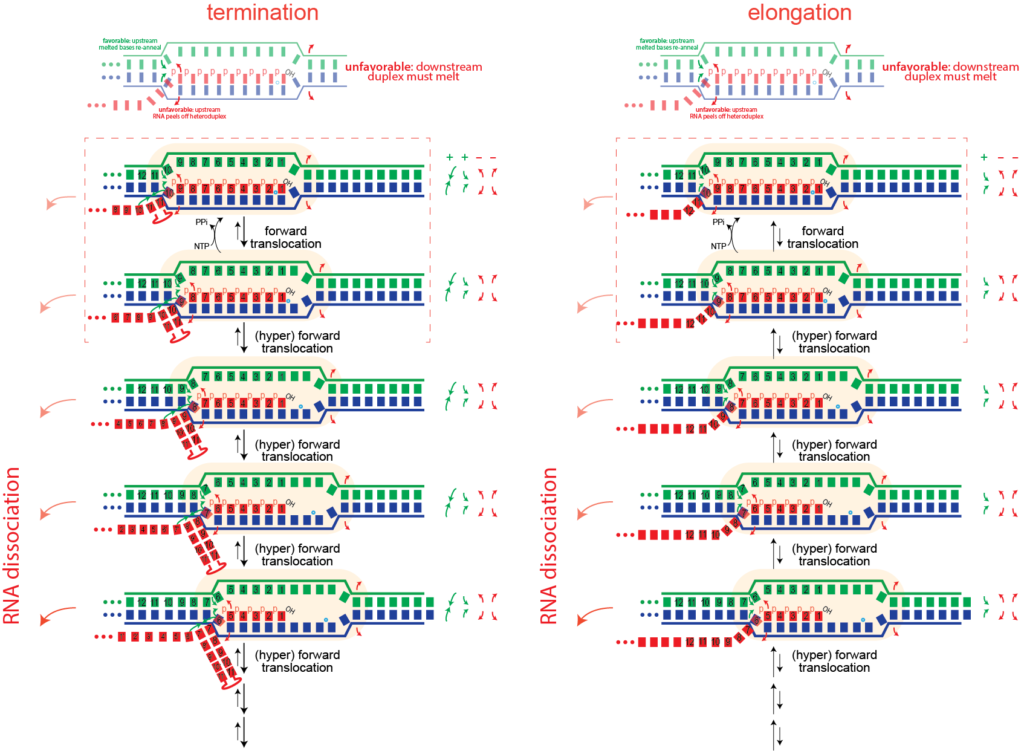
How does an elongation complex come apart in the middle of the DNA, as occurs during a process called termination?
For this we need to "unthread" the topological locking of the RNA around the DNA. We believe this happens through a process called (hyper) forward translocation.
RNA Polymerase is a Motor Protein - Forward Translocation. During normal elongation, there is a repeating cycle in which the enzyme adds a base to the growing RNA, then moves forward (forward translocates) along the DNA to prepare for the adding the next base (see review by Steitz). In the pre-translocated state below, the location for the incoming NTP is occupied by the last added base. To empty that spot, the enzyme moves forward by one base to the post-translocated state (or from the enzyme's view, the DNA and RNA have moved backward by one base.
Note that in the two step process above, both forward translocation and the substrate NTP binding are shown as equilibria (they are in thought to be in rapid Brownian exchange). Phosphoryl transfer, to add the new NTP to the RNA, can only occur from the post-translocated state, but when the bond forms irreversibly, it now prevents that Brownian exchange. The widely accepted model for the energetics of translocation involves this Brownian ratchet (a "power stroke" model is not needed here).
Binding of substrate leads to subtle, but significant motion of parts of the polymerase. In the pre-translocated state, the phenyl ring of Tyr639 stacks onto the last base pair of the RNA-DNA hybrid. Substrate binding requires displacement of this side chain, which also moves the helix to which Tyr639 is appended (the helix is a part of the "fingers" domain). That movement now positions the active site (at the very bottom of the picture above) for catalysis.
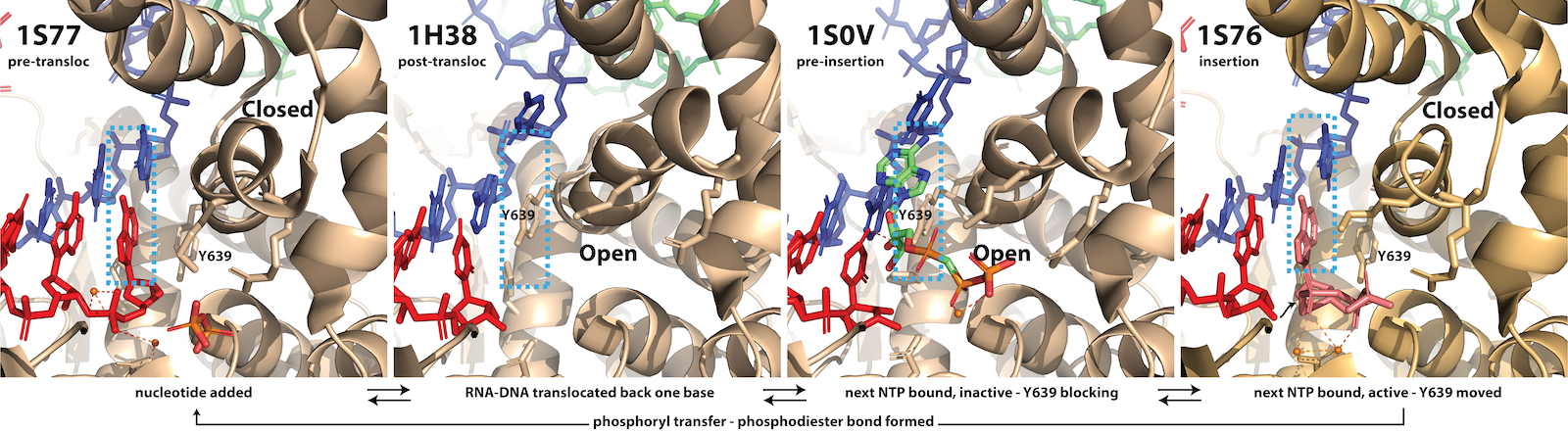
Fidelity of Base Incorporation. The third and fourth frames in the figure above illustrate a key feature of all polymerases in this larger family. The protein (C-terminal domain here) is comprised of fingers, palm, and thumb subdomains. The fingers subdomain can exist in an open or a closed configuration. Closing of the fingers domain positions key residues required for positioning of the substrate and the two essential Mg(II) co-factors for catalysis of the phosphoryl transfer that adds the incoming base to the growing RNA. (interestingly, in the structurally unrelated multi-subunit family of RNA polymerases (bacterial, eukaryotic) a domain known as the trigger loop plays the same role).
In the pre-translocated state, the just added base blocks binding of a subsequence NTP. Pyrophosphate release returns the fingers to the open configuration, which then allows forward translocation (of the enzyme along the DNA). This movement (which can equally be viewed as the nucleic acids moving to the reverse) generates the post-translocated state (in the fingers open configuration).
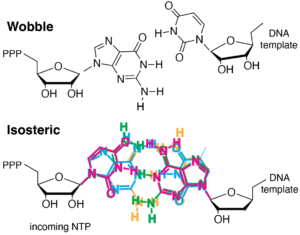
As noted, binding of the correct NTP (paired with the template strand base) allows for the fingers to close down around the active site. Proper closing of the fingers domain, which only occurs for an isosteric (Watson-Crick) base pair, is critical for formation of the catalytically competent phosphoryl transfer active site. The selectivity is critical for the fidelity of transcription. The figure below shows at least three stages at which elongation is regulated to achieve incorporation fidelity.
- For initial binding of the incoming NTP, an incorrectly paired base binds somewhat more weakly, so that the equilibrium lies more towards unbound (this is the most obvious, though perhaps less critical, requirement).
- Proper closing of the fingers domain to form an optimal active site is more favorable (only) with a substrate that forms an isosteric (Watson-Crick) base pair, such that for a mis-paired base, the equilibrium lies more towards the open configuration.
- In addition of poor structuring of the active site, an incorrectly paired (non-isosteric) base may have poor alignment for attack at its 5' phosphate, slowing phosphoryl transfer.
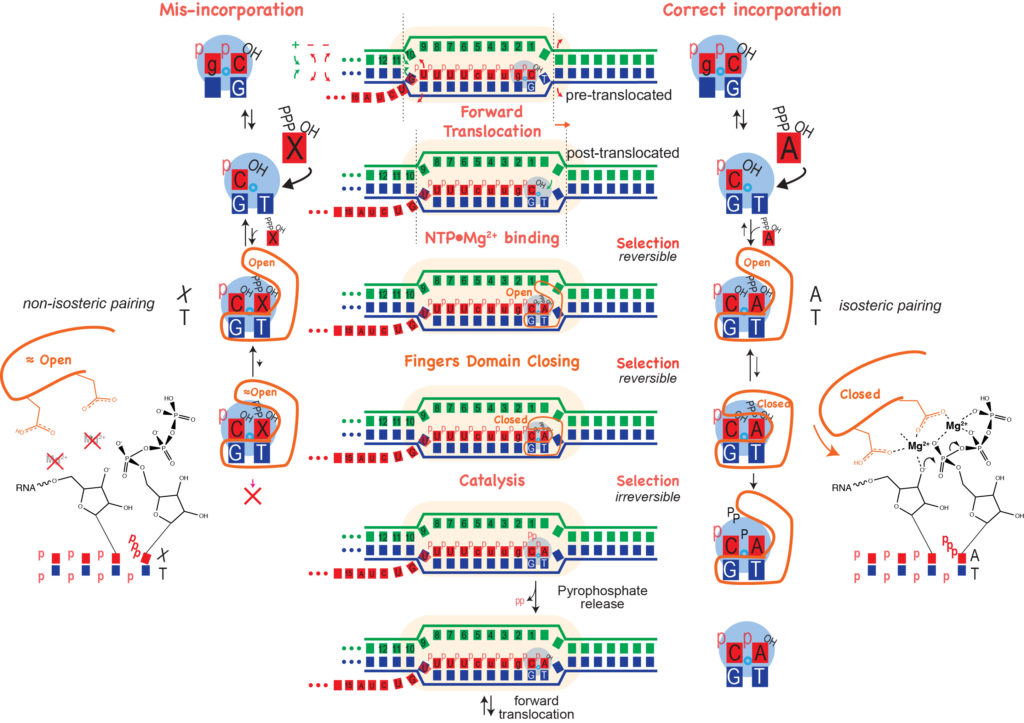
Polymerase mutants that favor closing of the fingers domain may actually transcribe "better," as shifting the closing equilibrium increases the population of the catalytically competent state, however this decreases the selectivity that ensures incorporation fidelity. In general, there is an inverse relationship between speed and fidelity - be careful what you select for!
From the literature (at least) the following mutations in the fingers domain have been shown to reduce T7 RNA polymerase fidelity: F644A, G645A, G640A, H784A (G640 and H784 are fully conserved, while F644 and G645 are largely conserved, across the family). G640 and F644 interact with the template DNA in the closed state. In the native enzyme, a non-isosteric base, binding at the active site via its triphosphate would be expected to "move"push" its partner template strand, interfering with proper closing of the fingers domain. Alanine substitutions might then soften that interface, allowing closing of the fingers, even in the presence of a mismatched base.
Hyper Forward Translocation. It is possible for the RNA polymerase to move forward more than needed, a process called hyper forward translocation. The second step above is hyper forward translocation. Both forward and hyper forward translocation involve the following energetic balance:
- melting of one base pair in the DNA (right-most red arrows)
- melting of one base pair in the RNA-DNA hybrid (left-most red arrows)
- reannealing of one base pair upstream
The first two above are unfavorable, balanced only partly by the third favorable process. Presumably, hyper forward translocation happens rarely under normal conditions. However, a "bumping" force from the left could be enough to drive this process. Similarly, we will see that formation of structure in the RNA as it exits the polymerase could also drive this process.
This one base pair movement is probably not enough to unthread the lock (otherwise undesired termination might happen during normal elongation). However, it does displace the 3' hydroxyl of the RNA from the active site; the polymerase cannot add another base until the complex reverse forward translocates back in register to re-position the 3' hydroxyl. With further hyper forward translocation, at some point, perhaps 4-5 hyper forward translocation events, the RNA may be sufficiently unthreaded to now be able to dissociate, allowing the bubble to collapse, releasing the polymerase.
Why doesn't this happen more often? Perhaps it does, but not with enough forward translocation steps to reach the point of critical instability. Note that each hyper forward translocation event is reversible - the polymerase can also reverse forward translocate and end up back in a configuration competent to continue elongation.
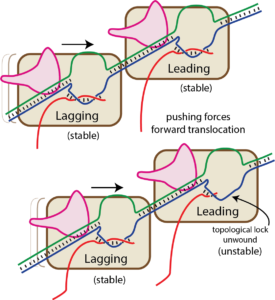
Bumping by a trailing (lagging) RNA polymerase can displace a leading RNA polymerase (this is true for the T7 family, but not for the multi-subunit family of RNA polymerases). We have proposed that this happens by the trailing RNA polymerase “pushing” against the leading RNA polymerase, which might lead to hyper forward translocation of the leading polymerase (as described above, hyper forward translocation reduces the size of the RNA-DNA hybrid and unthreads the topological lock). The resultant instability allows the complex to dissociate, ending transcription irreversibly.
As you can see in the "bumping" cartoon here, a trailing RNA polymerase can "push" a stopped RNA polymerase, sliding the stopped polymerase forward. But in order for that to happen, the melted bubble must move in accordance (the structure of the complex requires this).
Hairpin-dependent (Type I) termination. Another mechanism of driving hyper forward translocation is formation of structure in the RNA at the exit channel. Type I termination requires two things:
- a slowing of the RNA polymerase
- formation of structure in the RNA at the exit channel, such that structure formation "pulls" RNA out of the complex
Formation of structure just past (or in) the exit channel such that pulling more RNA out will allow more (stable) structure to form can also drive hyper forward translocation. The driving force here is different than bumping, but the effect is the same.
Note that this is inherently a kinetic competition. If the polymerase makes RNA faster than the structure forms, then sufficient RNA will be extruded from the active site to allow stable RNA structure formation without pulling on the RNA. Thus mechanist models that focus only on thermodynamic stability will not fully recapitulate termination.
Backtracking. Forward translocation that does not lead to termination, nevertheless forces the polymerase to pause until back translocation restores the 3' end of the RNA to its active site position. In principle, RNA polymerase can also backward translocate (back track) relative to that position. This will also remove the 3' end of the RNA from active site and lead to a paused state.
The above illustrates that in back tracking, the 3'-most base must dissociate from the template strand, while a hybrid base pair forms at the upstream edge of the hybrid (follow the numbering). Similarly, in the DNA, a downstream base pair forms as we melt a base pair upstream. While back tracking is common in the multi-subunit polymerases there was no evidence (until recently) that back tracking occurs in the single subunit RNA polymerases. There is no evidence that it does not...
Runoff transcription
Although RNA polymerase rarely encounters the end of a DNA duplex in vivo, this is the common form of synthesis in vitro. This process can also be viewed in the light of hyper forward translocation. More on this soon.
Content from earlier version of this web site
The RNA polymerase from bacteriophage T7 is the simplest and best-understood of the RNA polymerases. In a sense, it is the core of an RNA polymerase. As such, it represents an ideal model system in which to study transcription. It is less regulated than multi-subunit RNA polymerases and as a result, is more cleanly promoter specific.
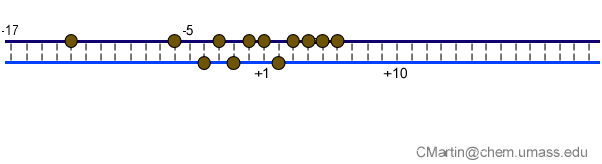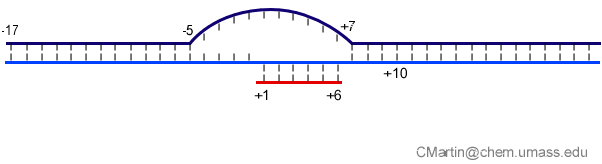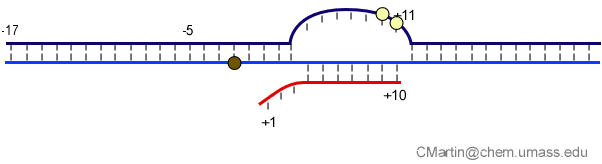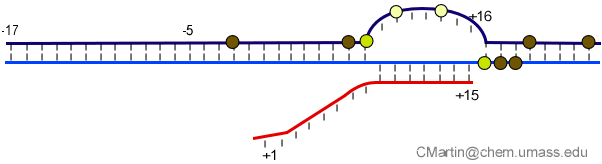
Fluorescence probes of melting. In order to follow the progress of the melted bubble during the various stages of transcription, we have used the site-specific incorporation of fluorescent nucleotide analogs into the DNA. In the summary below, dark circles represent fluorophores stacked within a duplex, while lighter circles indicate the increase in fluorescence associated with melting of the DNA bubble.
The initially melted bubble is about 8 bases in size [xx]. As transcription initiates, the upstream edge of the bubble remains fixed as the downstream edge expands [xx]. Just past the synthesis of an 8 base RNA, the upstream edge of the bubble collapses, ultimately returning the bubble to the constant 8 base size characteristic of an elongation complex [33], clear of the promoter.
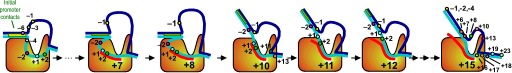
A large structural change in the protein. The structures above reveal a very large structural rearrangement of about 1/3 of the protein. Based on topological and biochemical considerations, we have developed a movie to illustrate how we think this transition occurs.
{kind=link}
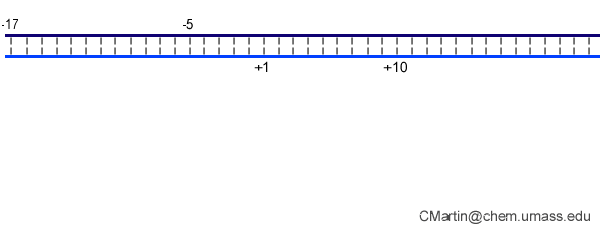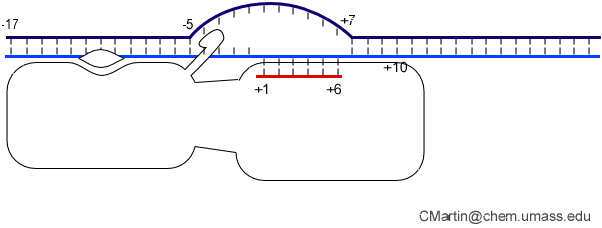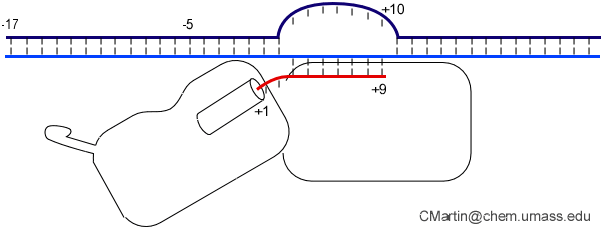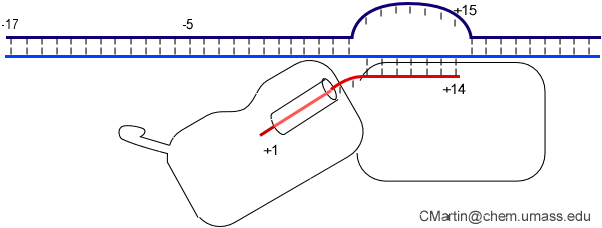
Biochemical studies complement the fluorescence studies, revealing precisely when promoter contacts are lost and when the 5' end of the RNA is displaced from the template. The cartoon above shows details of a functional model we have proposed (see also our animation combining the structural and biochemical models). In particular, we argue that binding of the promoter DNA drives the Val loop into the DNA, initiating bubble formation. the structural movie shows that the promoter-protein contacts can be retained (and therefore the bubble can remain open) through translocation to about position +8, consistent with our biochemical studies. At that point, however, the specificity loop is pulled from the promoter-binding platform, releasing the DNA. This in turn, removes the Val loop from the upstream edge of the bubble, allowing collapse. Collapse then helps to drive initial displacement of the 5' end of the RNA, leading it into the just-formed RNA exit channel. Threading of the RNA into the channel signals the formation of a stable elongation complex.
{kind=link}
The following describes recent advances from our laboratory.
View animated cartoon in: QuickTime | Flash | GIF format
{kind=link}
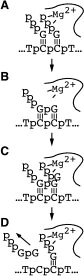
Abortive cycling and promoter clearance. Our very recent work has turned towards understanding the RNA polymerase as it transcribes away from the promoter. Fluorescent base analogs have proven extremely valuable in this analysis and we have extended our earlier characterization of the initial promoter complex to provide an accurate mapping of the downstream edge of the bubble [unpublished]. Additionally, we have shown that in the synthesis of a trinucleotide transcript, the downstream edge of the bubble does not move the active site approaches very close to the downstream edge of the melted region, a result
Available crystal structures are consistent with earlier footprinting studies in that the RNA polymerase can transcribe at least 3-6 bases without releasing the upstream duplex promoter contacts. Modeling from the crystal structure, however, predicted that during this early phase, the heteroduplex can be no longer than 3 base pairs. Using fluorescent base analogs, we have mapped the collapse of the initial bubble and the extent of heteroduplex formation [37]. The results show very clearly that the initial heteroduplex grows to a maximal length of about 10 base pairs. On translocation from 8 base pairs to 9 or 10, the initial melted bubble collapses, indicating the loss of promoter contacts, and on translocation from position +10 to +11, the initially synthesized RNA first begins to peel away from the heteroduplex. These results have important implications on the mechanism driving the structural changes occurring at this unique stage in transcription.
Probe development. A necessary component of the above analyses was the advent of fluorescent base analogs beyond the initial analog of adenine, 2aminopurine. In the above work, we have used two new analogs: 6MI, an analog of G (collaborative with M. Hawkins, NCI) and pyrroloC, an analog of C (in cooperation with J. Randolph and H. Mackey, Glen Research Corp.), all of which show duplex-enhanced fluorescence quenching). This has extended the sequence contexts which can be probed (a T analog, furanoT, will be characterized in the near future). These new probes have excitation maxima more distant from protein absorbances, reducing greatly complications of background fluorescence. We are currently collaborating with J. B. A. Ross (Mt. Sinai) to use lifetime measurements as a means of characterizing translocational heterogeneity in various stalled complexes, to clarify further the picture above. Finally, we have demonstrated the use of mismatch probes to clearly distinguish DNA:DNA from RNA:DNA duplexes [37]. These new tools and approaches will likely be of utility in a wide variety of protein-nucleic acid systems.
Site-specific de novo initiation. Template strand DNA directs the synthesis of RNA and is clearly essential for polymerase function, however, we have demonstrated that start site selection is not strongly dependent on the precise nature of the coupling between the non-transcribed and the transcribed regions of the template strand [29]. This has led to more recent studies (with W. T. McAllister) which strongly favor a model in which the linkage to the upstream domain serves merely as a tether to increase the local concentration of an appropriate initiation sequence near the active site [35], despite the appearance in the crystal structure of what appears to be active guidance by the linking DNA.
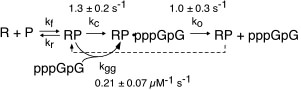
Kinetic studies of initiation. As described above, our early work used steady state kinetics to assess functional structural elements of promoter binding. We subsequently turned our attention to pre-steady state kinetic analyses of initial dinucleotide synthesis in order to elucidate the mechanism of initiation [32]. Fitting kinetic data to integrated rate equations for specific, complex mechanisms, we have demonstrated that dinucleotide re-binding (product inhibition in this initial assay) must be included in any such mechanism and that substrate activation plays an important role in mediating product release. These results present an essential foundation to mechanistic studies of translocation through the initial abortive region, leading to promoter clearance.
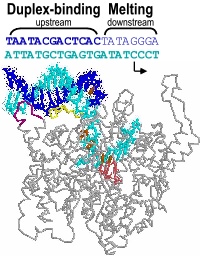
Promoter melting. Two unique roles of an RNA polymerase are promoter melting and the de novo (without a primer) initiation of RNA synthesis. With respect to the former question, we have used fluorescent base analogs to characterize the initially melted bubble, mapping the melted region (the second domain described above) and demonstrating that promoter melting is extremely rapid, suggesting that melting occurs coincident with initial promoter binding [28]. The latter result was expanded in thermodynamic measures of the binding of full length and truncated promoter constructs [30]. The observation that truncated DNA constructs representing the duplex binding domain (only) bind to the protein more strongly than full length DNA led us to a model in which the unfavorable process of melting is obligatorily coupled to binding. Our gel-based bending assays [31] support this notion and have suggested a structural mechanism in which binding-induced bending of the DNA leads to melting, a model which we are currently testing by site-directed mutagenesis.
Promoter recognition and binding. Our early research (1988-1996) employed functional group substitutions in DNA to characterize promoter contacts and interactions in the T7 RNA polymerase model system [21, 22, 25, 27]. The results are consistent with a two-domain description of the promoter and predicted energetically critical major groove contacts in the central part of the proposed upstream duplex binding region of the promoter, with less important minor groove contacts in the far upstream part of the bound duplex. Our results further showed that within the melted domain encompassing the start site for transcription, the nontemplate strand provides few energetically important contacts [23]. These results are fully consistent with the subsequent (1999) crystal structure of the promoter bound complex (and with a large body of full base pair substitution measurements), and provide energetic information not available from the structure alone.
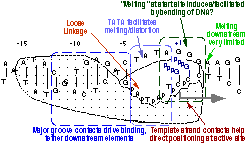
We have confirmed a central "core" within the promoter which is responsible for tight binding to the RNA polymerase. Energy from this interaction appears to be used to drive melting of the DNA near the start site. Other recent results have yielded unexpected (and therefore very intersting!) results, providing information on how the polymerase directs the initiating bases of the DNA template strand into the protein active site. Click here (or on the picture above) for more detailed information.